
羟醛反应
| 羟醛反应 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 反应类型 | 偶联反应 | ||||||||
| 反应 | |||||||||
| |||||||||
| 反应条件 | |||||||||
| 温度 | -Δ, ~-70°C[a]
| ||||||||
| 催化剂 | -OH或H+
| ||||||||
| 标识 | |||||||||
| 有机化学网站对应网页 | aldol-addition | ||||||||
| RSC序号 | RXNO:0000016 | ||||||||

典型的羟醛缩合反应

典型的现代羟醛加成反应如上图所示,即酮的烯醇负离子对醛的亲核加成。反应后,羟醛产物在一定条件下可以失一分子水形成α,β-不饱和羰基化合物,这时的羟醛反应及脱水过程可称「羟醛缩合反应」。在羟醛反应中可以参与反应的亲核试剂有:烯醇、烯醇负离子、酮的烯醇醚、醛和其他羰化物。与之反应的亲电试剂通常是醛或酮(包括许多反应变种,如曼尼希反应)。若亲核试剂和亲电试剂不同,反应称「交叉羟醛反应」;若亲核试剂和亲电试剂相同则称「羟醛二聚反应」。

发现与发展历程
[编辑]羟醛反应首先由法国化学家查尔斯-阿道夫·武尔茨[8]和俄国化学家亞歷山大·波菲里耶維奇·鮑羅丁[9][10]于1872年分别独立发现,最初以氢氧化钠為乙醛加成反应之条件,产物是有羟基的醛化物,羟醛即由此得名。由于羟醛反应的产物控制方法学还未出现,交叉羟醛反应总会产生大量无任何合成价值的副产物,除了利用简单醛酮分子的羟醛缩合反应可用于合成共轭不饱和醛酮之外,该反应在发现后的近一个世纪内一直没有廣泛用處[11]。
羟醛反应第一件具里程碑意义的事件出现于1957年,当时美国西北大学的H.E.齐默曼(Zimmerman)和M.D.特拉克斯勒(Traxler)为解释格氏试剂介导的伊万诺夫反应中反式产物占优势的问题,提出了著名的六元环过渡态模型,后人常称之齐默曼-特拉克斯勒模型。[12]该模型首次从立体化学角度剖析羟醛反应,指出烯醇盐构型与产物立体化学之间的对应关系,成为羟醛反应历史上的第一个理论突破并在很长一段时间内成为后续研究的关键指导理论。以后众多的实验结果也证明这模型的準确度,依据模型设计的实验大多得到模型预期的立体化学结果[13]。
二十世纪六十年代,核磁共振和X单晶衍射技术大力推动了立体化学的发展[14],也为羟醛反应研究带来了极大便利。通过氢谱中获得的大量积分、化学位移、偶合常数等数据,可方便快捷地分析鉴定产物的立体化学结构;X单晶衍射可以通过晶体衍射来分析化合物的绝对构型,而在此背景下羟醛反应的研究也开始逐步升温[15][16][17]。
1973年日本北里大学教授向山光昭发现了用硅醚形态稳定烯醇以促成羟醛反应的新方法[18][19],该发现为羟醛反应研究揭开新一页。向山羟醛反应通过预先制备烯醇硅醚与醛酮混合后在路易斯酸催化下反应得到羟醛产物。由于产物的立体化学与烯醇硅醚的构型之对应关系不再符合齐默曼-特拉克斯勒模型,因此提出向山开链过渡态理论以解释相关反应结果。通过路易斯酸在向山羟醛反应中的研究,羟醛反应立体化学控制技术得以发展,并成为该羟醛反应立体化学三大控制方向之一[20][21]。
二十世纪六十年代到八十年代是有机化学发展史上的重要时期,出现了众多不对称合成技术和著名的有机化学家,如:野依良治,巴里·夏普莱斯和威廉·斯坦迪什·诺尔斯。羟醛反应立体控制技术同样在这段期间发生了重大的突破[22]。1981年哈佛大学教授大卫·埃文斯[23][24]发明了手性噁唑烷酮配体介导的不对称羟醛反应技术,第一次实现高对映选择率的不对称羟醛反应。此外日本的向山光昭、克里明斯等都在这段时间在羟醛反应立体控制技术有重大贡献。
近年来,化学家对于羟醛反应的研究集中在催化剂的发现领域[25]。许多更新颖的催化剂出现,包括酶类与抗体型催化剂[26]:pp. 273–310[27][28][29][30][31][32][33]、手性金属络合物催化剂[34],以及众多小分子催化剂[35][36]。
有机合成中的地位和意义
[编辑]羟醛结构单元存在于许多分子(包括天然产物和合成分子)中[37][38][39]。例如,通过羟醛反应大规模合成的日用化学品季戊四醇[40]及心脏病药物阿托伐他汀[41][42][43]。羟醛反应之所以应用广泛,是因为它将两粒相对简单的分子结合成一粒较复杂的分子,且通过形成两粒新的手性中心(于羟醛产物的α-碳原子上,在下述分子式当中标注)使分子更复杂。现代化学方法学不仅可以做到高收率的羟醛反应,而且能够控制反应产物的相对和绝对立体化学构型(見下文立體選擇性一節)。选择性合成特定的手性异构体对于现代药物化学非常重要,因为不同的手性异构体可能有完全不同的临床药理学特性[44]。
手性羟醛单元在聚酮化合物中较常见,聚酮是在生物有机体发现的分子。在大自然,酶催化聚酮發生多重克莱森缩合反应。这些反应产生的1,3-二羰基化合物可衍生出各式各样有趣的结构,其中一些有强效生物活性,如强效免疫抑制剂他克莫司[45][46]、抗癌药物盘皮海绵内酯[47][48]及抗真菌药物两性霉素B[49][50]。不对称羟醛反应在合成方法学的应用,使许多传统的天然产物分子合成成为可能,且在许多合成中成为关键步骤,如合成littoralisone[51],福司曲星[52][53]及callipeltoside[54][55]。
反应机理
[编辑]羟醛反应的具体机理还不确定,但现今广泛认可的机理有两种[56][57][58][59]:羰化物如醛或酮可转化为烯醇或烯醇醚。它们的α-碳原子都亲核,可以进攻活泼的质子羰化物,如质子化醛,称“烯醇机理”[60][61]。
羰化物或碳原子有活泼氢的有机物,可于羰基α位去质子形成烯醇负离子,而该离子形态比烯醇和烯醇醚更亲核,可直接进攻亲电试剂。常见的亲电试剂为醛类化合物,而酮活性较低,这类反应机理称“烯醇负离子机理”[62][63][64]。
若反应条件特别剧烈,如:甲醇钠作碱,甲醇为溶剂的回流条件下,则会发生缩合反应;为了避免这种情况可以降低温度并使用温和碱如二异丙基氨基锂,并使用四氢呋喃为溶剂,在−78 °C反应。虽然羟醛加成反应通常能完全反應,却并非不可逆。用强碱处理羟醛加成产物,可逆向裂解羟醛而得到起始原料。羟醛缩合通常认为不可逆,但交叉羟醛反应动力学研究表明其实际可逆[65]。

羟醛反应的概括

烯醇机理
[编辑]在酸催化条件下,反应机理的首步是酸催化羰基异构成烯醇,并加质子啟動另一分子羰基,使其高度亲电。烯醇的α碳原子亲核,能进攻质子化羰化物,而后脫质子形成羟醛。羟醛产物最后通常还会继续脱水得到不饱和羰化物[66]。以下展示典型酸催化醛自身缩合反应[67]:
酸催化羟醛反应机理
[编辑]
酸催化脱水
[编辑]
烯醇负离子机理
[编辑]若催化剂是温和的碱,如氢氧根离子或醇负离子,则羟醛反应可通过形成共振稳定的烯醇负离子而亲核进攻另一分子羰化物,形成羟醛产物醇盐,而后自身脱水得到不饱和羰化物[68]。反应式展示了碱催化醛自身羟醛反应的机理[67]:
碱催化羟醛反应
[编辑]图例使用−OCH3做碱

碱催化脱水
[编辑]此反应通常错写为简单一步,见E1cB消除反应

某些情况下反应仅需要催化量的碱,但大多数反应都需要等当量的强碱介导反应,如二异丙基氨基锂(LDA)或二(三甲基硅基)氨基钠(NaHMDS)。这情况下烯醇负离子的形成是不可逆的,直到羟醛产物的金属醇盐在酸化处理后质子化,羟醛产物才得以形成。[69]
齐默曼-特拉克斯勒反应过渡态模型
[编辑]有机反应过渡态是控制反应的关键要素之一。羟醛反应的第一个过渡态模型是1957年由霍华德·齐默曼和马乔里·特拉克斯勒提出的六元环过渡态模型[12],即如下图所示齐默曼-特拉克斯勒模型,M是由碱带入的金属离子。运用建立在环己烷上的构象分析理论可以看出,醛分子在受烯醇进攻时的构象形态主要是使羰基两侧较大的取代基处于六元环平伏键的位置,因为可以使各官能团之间空间位阻最小,过渡态的能量也最小,而导致优先形成相应立体构型的产物。[70]

齐默曼-特拉克斯勒模型

从图中同样可以看出,α-碳上的立体构型由烯醇盐的构型决定。Z型烯醇主要生成顺式羟醛,而E型烯醇生成以反式羟醛为主的产物。值得注意的是,上面的顺反构型是相对构型,因为存在两种顺式产物和两种反式产物。换言之,上面每个产物对应两种可能的绝对构型。[71]
交叉羟醛反应的反应物控制
[编辑]羟醛反应的一个主要研究方向是如何“控制”反应产物,如以下假定反应的两粒不对称酮以乙醇钠為条件缩合。乙醇钠的碱性不能将其中任意一粒酮的邻位氢完全脫去,但可以将两种酮都形成少量的烯醇钠盐。这意味着两种酮分子不仅都是潜在的亲电试剂,而且都可以通过形成烯醇负离子成为亲核试剂。最后反应的结果是两种亲电试剂和两种亲核试剂形成的四种产物[72]。

四种可能的羟醛反应产物

所以,若希望得到交叉羟醛反应-产物中的一种就必须严格的控制其中一个羰基分子成为亲核试剂,另一个羰基分子保持亲电的分子形式。[73][74]
酸性
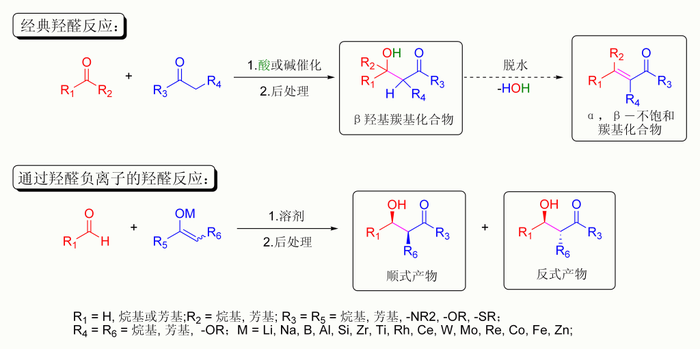
[编辑]最常用的控制方法是:两种参与反应的羰基分子中只有一种具有α氢原子,而只有这种分子才能异构化成为烯醇式成为亲核试剂。例如,丙二酸酯和苯甲醛反应则只会形成一种产物。因为只有丙二酸酯具有α氢原子而成为亲核试剂,不能进行烯醇化的苯甲醛则只能成为亲电试剂:[75]
羟醛反应的酸性控制

丙二酸酯非常易脫质子形成烯醇负离子,因为其羰基α位连接另外一組羰基。1,3-二羰基形式能让形成的烯醇负离子更稳定,所以不需要很强的碱就能烯醇化。此效应可以引申出另外一种情况:即使羰基旁有两种α氢原子,却仍然可以控制形成哪种烯醇式。若两种α氢原子酸性差异夠大,碱就会中和较酸的质子形成烯醇,而酸性较弱的α氢原子则不受碱影响[76][77]。这种控制方法,只存在于酸性差异够大而碱不过量的情况下。其应用在亚甲基临近于两組羰基或者氰基的分子中较常见,如Knoevenagel缩合反应和丙二酸酯合成的第一步[78][79][80]。
加料顺序
[编辑]另一种常用控制选择性的解决方案是先让其中一種羰化物形成烯醇负离子,然后在动力学控制下加入另一羰化物。[81]动力学控制决定了正向羟醛加成反应必须明显快于反向羟醛反应。要达成这目的必须满足其他两項条件:1、量化形成其中一种烯醇负离子;2、正向羟醛反应明显快于烯醇离子的交换过程。常见的动力学控制操作是使用二异丙基氨基锂在−78 °C下和酮反应得到烯醇负离子,而后缓慢加入醛[82]。
烯醇和烯醇负离子
[编辑]互变异构
[编辑]烯醇是醛或酮的异构体[83]。在存在少量酸或碱的条件下烯醇和醛酮可以相互转化,属于一种互变异构[84][85][86],或称之为烯醇和醛酮互为互变异构体。酸性或中性条件下烯醇不稳定,互变异构的反应平衡有利于醛酮而不利于烯醇,所以醛酮中只含有微量的烯醇异构体[87]。如下图所示:

形成
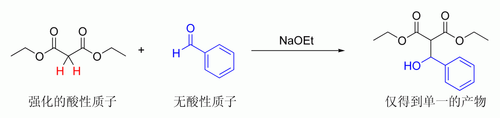
[编辑]相比酮式,通常烯醇式是一种不稳定的互变异构形态,有三种较稳定的烯醇化合物:一、形成烯醇的双键与分子内其他双键共轭[88]。二、形成的烯醇式与较大的芳香基团连接[89]。三、形成的烯醇式的双键碳原子直接连接多粒氟原子[90]。而烯醇负离子可通过两种方法获得:1、强碱(“硬条件”) 2、路易斯酸和弱碱(“软条件”)[91]。

要脫質子,立体电化学需要α-碳-氫σ键必须能够和羰基π轨道重叠[92]。
去质子化需要的反式共平面

立体化学
[编辑]烯醇化的区域选择
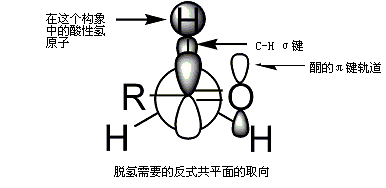
[编辑]如果不对称酮与碱反应,有生成两种「区域异构」烯醇的倾向(忽略烯醇本身的立体化学)[來源請求],如:
动力学和热力学控制的烯醇

三取代烯醇认为是「动力学控制」的烯醇,而四取代烯醇则认为是热力学控制的烯醇。α氢原子脫质子后形成的动力学烯醇相对位阻更小,脫質子更快。总体来说,四取代烯烃由于超共轭,比三取代烯烃更稳定。烯醇区域异构体的比例很大程度受反应中碱的影响。以上述为例,动力学控制可以通过二异丙基氨基锂在−78 °C下得到,且有99:1的动力学產物;而热力学烯醇可以通过三苯基甲基锂在室温得到1:9的热力学產物[93]。一般来说,动力学烯醇更倾向于在低温下形成相对离子化的金属-氧键,而反应中快速脫質子須使用略过量的碱,该碱需是强且高位阻的碱。体积大的碱只会中和位阻小的氢原子,低温条件和过量的碱能防止在初步烯醇化时平衡转化为更稳定的烯醇式。热力学烯醇更倾向于长的平衡时间与更高温,这种条件形成了相对共价的金属-氧碱而且使用少量强碱。不足量的碱能同时脫去所有羰基分子的质子,不同的烯醇负离子和羰基之间交换质子并形成平衡,从而形成更稳定构型。这种选择可以由挑选金属离子和溶剂来控制[94]。
关于不同条件下烯醇化的扩展研究已经开始。当代有机化学在大多数情况下,已经可以得到想要的烯醇构型:[95]

烯醇生成的立体选择性

烯醇化方法
[编辑]醛酮羰基的α氢pKa一般为23左右。为了避免自身发生羟醛加成则需要使用如二异丙基氨基锂的强碱,才可迅速地使整个反应体系完全烯醇化。由于使用强碱,且提高脫質子的选择性,通常在丙酮乾冰浴的低温下反应,溶剂则通常用四氢呋喃、二氯甲烷等非质子极性溶剂。由于二异丙基氨基锂(LDA)这类强碱属于硬碱,所以这种方法属于硬式烯醇化。如果使用路易斯酸先与醛酮羰基络合,则能极大地提高α氢的酸性,就可以使用诸如三乙胺的弱碱反应,从而实现脱质子烯醇化,这种方法就属于软式烯醇化[96]。
烯醇盐的顺反异构
[编辑]烯醇或烯醇盐相当于烯烃的衍生物而有顺反异构体。一般用E表示反式,Z表示顺式。[97]见图:

该烯醇的命名法与烯烃体系有区别,规则是:只要是羰基氧原子烯醇化后与双键另一端的大基团处在一端的都认为是顺式,而与另一个R'的基团大小无关[98]。如:

图例之烯醇盐按照普通规则属反式,因硫的原子序数比氧大而基团优先,但在羟醛反应的研究中属顺式。而对于酮化物,大多数烯醇化条件都是得到Z烯醇。而对于酯化物,大多数烯醇化条件得到的是E烯醇。而在加入六甲基磷酰胺后,该脫質子过程的立体选择性可以与上述相反[99][64]。

烯醇形成的立体选择性可用Ireland模型解释,[100][101][102][103]虽然这种模型的准确度还未確定,如在大多数情况下无法得知在中间体是单体或是低聚体;即使如此Ireland模型仍然是理解烯醇化过程的有用工具。[104]

Ireland模型

Ireland模型中,去质子化过程可能有六元环过渡态。两个亲电取代基团当中偏大的基团(在例子当中,甲基比氢原子大)在过渡态中倾向于采取平伏键的位置,导致优先得到E构型的烯醇。在以下情况模型失效:溶剂体系从四氢呋喃变为23%的六甲基磷酰胺-四氢呋喃会造成烯醇的立体化学发生逆转。[105]
立体选择性
[编辑]羟醛反应非常重要,因为其过程能生成两粒手性中心。许多相关研究已经了解反应机理并能通过不同的条件提高反应选择性。顺反转化通常使用α和β碳原子上的相对构型表示。

文献也用过赤式/苏式来命名一些碳水化合物的立体构型。而有机化学家们一直感兴趣的问题是如何控制羟醛反应,使四种可能产物中的一种成为主要产物,并尽量减少其他异构体的生成。
烯醇双键构型的影响
[编辑]每一个烯醇的双键构型都确定了主要产物的相对立体构型,E型双键得到反式产物;Z型双键得到顺式产物:[95]

双键构型对产物顺反异构的影响

烯醇盐的顺反异构直接影响羟醛产物的立体化学,故須控制烯醇盐的顺反异构以期获得特定结构的烯醇盐。在胺类碱作用下,烯醇化的结果倾向于生成反式烯醇盐,如果在反应体系内添加六甲基磷酰胺(HMPA),则可以使上述结果相反。此现象可用「Ireland模型」解释:

生成反式烯醇盐的六元环过渡态大取代基间相互积压程度小、能量低,容易形成过渡态,进而得到反式烯醇盐;同理,生成顺式的过渡态存在大取代基1,3-竖键相互左右,挤压程度高、能量上不利于形成过渡态,故顺式含量低。
羟醛反应研究的发展产生了更为可靠的烯醇化技术,通过选择不同的碱和路易斯酸催化剂,可以有效地产生97%以上的单一烯醇盐。基本上而言,使用小位阻硼硬路易斯酸三氟甲磺酸二正丁基化硼(Bu2BOTf)和大位阻碱N,N-二异丙基乙基胺(DIPEA)有利于生成顺式烯醇盐;反之,大位阻软路易斯酸氯代二环己基硼烷(Cy2BCl)和小位阻碱的搭配则有利于生成反式烯醇盐。见下图:
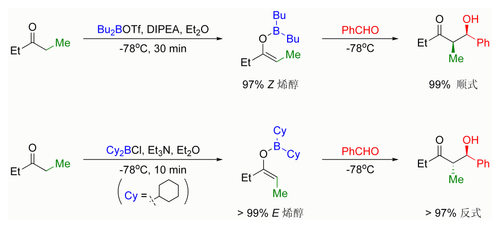
位阻对于立体选择性的影响
金属离子的影响
[编辑]烯醇金属离子的在确定羟醛反应的立体选择性上有很重要作用。硼试剂[106][106]就常用于选择羟醛反应的立体產物,因为其键长远比其他金属离子(如锂,铝或者镁)短。
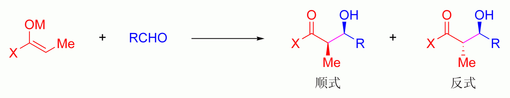
硼代替金属离子对于立体选择性的影响

硼-碳和硼-氧键长分别为1.4–1.5Å和1.5–1.6Å,而典型金属-碳和金属-氧键长分别为1.9–2.2Å和2.0–2.2Å。硼试剂让金属原子“收紧”过渡态而让反应有更高的立体选择率。[107]这样,上述反应使用烯醇锂负离子得到的顺反比为4:1;而使用二丁基硼烯醇则得到97:3的高选择性。
烯醇α-手性中心的影响
[编辑]羟醛反应可发生“受質介导的立体化学控制”,也就是反应受質的手性可影响产物的手性。若烯醇受質有α位手性中心,则可完美控制立体化学。

基于烯醇立体化学控制的羟醛反应

E烯醇之主要控制因素为1,3-烯丙位张力;而Z烯醇,主要控制因素则是防止1,3-位的双直立键相互作用。总模型如下:

基于烯醇立体化学控制的羟醛反应总模型

为了让图解更清晰易懂,烯醇分子的立体化学被差向异构化;而在实际反应中,醛的差向异构面也可发生亲核进攻反应。两例中,1,3-顺式的非对映体都有利。此类立体化学控制的例子有许多:[108]

亲电试剂α手性中心的影响
[编辑]当烯醇进攻的受質醛有α位手性中心,同样可以完美控制立体化学。大多数的研究表明E式烯醇参与了Felkin非对映体的选择;Z烯醇参与反Felkin的选择性。总模型[109][110]如下所示:

羟醛反应的立体化学控制的总模型

由于Z烯醇必须通过有不稳定的顺戊烷中间体或反Felkin旋转异构体來反应,所以Z式烯醇在这例子中降低了非对映选择率,如下:[111][112]

烯醇构型对立体化学控制的影响

立体诱导的统一模型
[编辑]若烯醇和醛分子本来有手性,则可使用统一的立体化学模型以预测该“双重手性区分”的羟醛反应,而该模型需要同时考虑两个因素:烯醇分子的空间位阻和立体化学的对于反应的影响,以及醛的空间位阻对于反应的影响。[113]一些关于此模型的应用如下所示:[112]

立体诱导统一模型

埃文斯噁唑烷酮化学
[编辑]现代有机合成化学需要合成光学纯的化合物。然而羟醛反应生成两粒手性中心,即形成四种手性异构体:

羟醛反应形成的手性异构体

现今已发展出许多用于控制相对手性(如顺或反)和绝对手性化学(如R S构型)的方法。

绝对手性控制与相对手性手性控制的关系

一種广泛使用的方法为埃文斯酰基噁唑烷酮法,[23][24]由大卫·埃文斯和同事于二十世纪七八十年代发现,这种方法通过添加手性助剂来暂时建立手性烯醇。这手性助剂通过非对映选择性反应将“手性”转移至产物,之后脱除助剂,得到需要的對映体。

埃文斯手性助剂的原理

在埃文斯的方法中,引入的手性助剂为噁唑烷酮,而形成的羰化物是一种酰亚胺。许多噁唑烷酮试剂现在都已商品化,且可买到两种对映体,其售价约每克10至20美元。
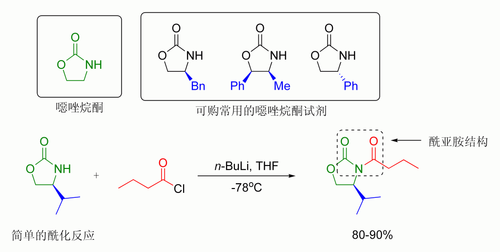
噁唑烷酮作为手性助剂参与不对称合成

噁唑烷酮的酰化反应操作过程简单。Z型烯醇可以通过「硼介导软性烯醇化」得到顺羟醛加合物。[114]
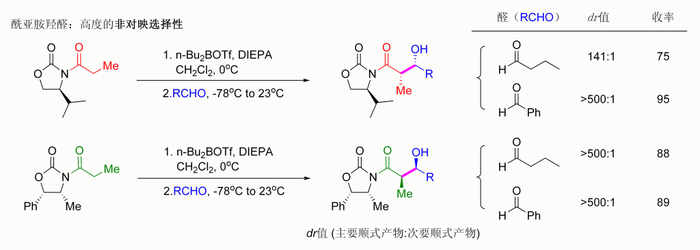
高非对映选择性的埃文斯羟醛反应

通常单種非对映体可以羟醛加成物结晶获得。尽管成本高且只能得到“顺”产物,埃文斯羟醛可靠,用途广泛。一些断裂助剂的方法如下:[115]

埃文斯手性助剂断裂法

构建酰亚胺结构都可發生选顺和選反羟醛加成反应,其允许形成四种当中三种手性组合:选顺[116]和选反:[117]

埃文斯羟醛反应形成的不同手性组合

在选顺反应中,两种烯醇化方法都如预期的得到Z烯醇,然而反应的手性化学并非受到噁唑烷酮而是甲基手性中心控制。该法还允许选择组建聚酮的手性(一类有反合成子的天然产物)。
现代方法与变化
[编辑]羟醛反应的现代化学方法学发展出了许多不对称羟醛反应,这些不对称反应常使用催化量的手性配体。当反应使用少量的光学纯配体,即可诱导形成光学纯的羟醛反应产物,该反应也属于一种“催化的非对称”反应。根据催化剂的不同可以将非对称反应分类为以下几种羟醛反应类型:
乙酰羟醛反应
[编辑]前文所述手性助剂主要局限在于N-乙酰酰亚胺反应無选择性。早期解决方法是暂时引入硫醚基团:[115][c]
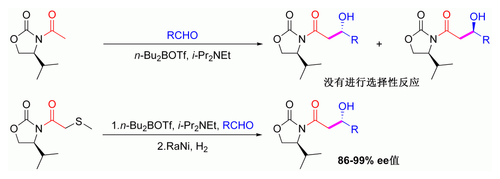
硫醚介导的乙酰羟醛反应

向山羟醛反应
[编辑]向山羟醛反应是在路易斯酸,如三氟化硼或四氯化钛的催化下,硅烯醇醚对醛的亲核加成反应。[118][119]向山羟醛反应并不符合齐默曼-特拉克斯勒模型。后来卡雷拉(Carreira)发现了一种实用的利用硅烯酮缩醛的不对称合成法:使用含钛的西弗碱催化反应(见下图)。这种催化剂有高度的对映选择性及广泛的受質适用性,在优化条件下,只需0.5%摩比的催化剂便可以得到高收率且高對映體過剩(ee)的加成产物[120][121][122]。该方法常应用于非支链脂肪族醛,由于在对映面两侧的电性和手性差别较小,该受質的亲电性对于催化不对称反应的通常较弱。[123]

向山羟醛反应

插烯物的向山羟醛过程还可以发生催化非对称性反应。下图展示了仅对芳香醛的有效反应,认为其机理与金属键合的手性二烯醇有关。[124][125]
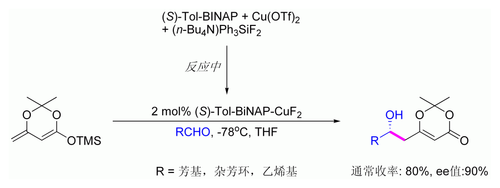
乙烯类似物向山羟醛过程

可理民斯噻唑硫酮羟醛反应
[编辑]近期[何时?]发现的埃文斯助剂按其发现化学家命名为可理民斯(Crimmins)噻唑硫酮催化剂[126][127],虽然不及埃文斯的反应实例中的数据,但这种催化剂能够使反应的收率、非对映选择性和对映选择性都大大提高。而优于早期的埃文斯助剂,噻唑硫酮对于乙酰羟醛反应同样有效,[128]且克里明斯发现可以通过使用少量的(-)-金雀花碱得到“埃文斯顺式”或“非埃文斯顺式”的加成产物。该机理认为是形成钛-键合六元环过渡态反應,而该过渡态机理类似于埃文斯助剂。
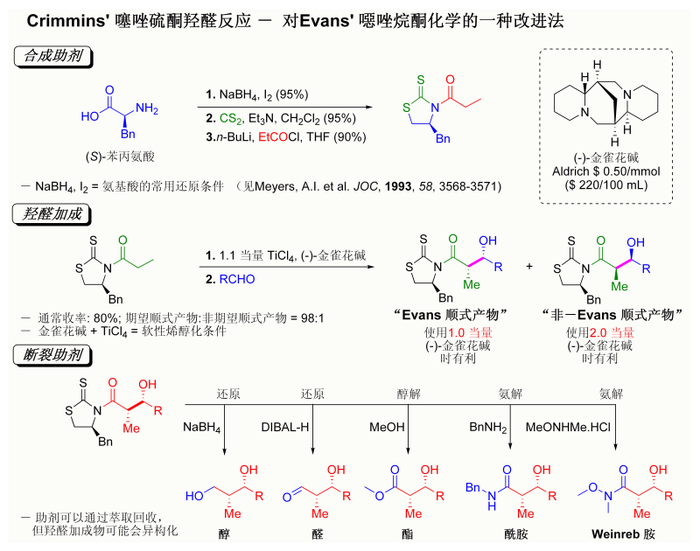
克里明斯噻唑硫酮羟醛反应

有机催化法
[编辑]羟醛反应的另一最新进展发现了可用手性二级胺催化反应。这种二级胺和酮反应会先形成烯胺中间体,并与合适的醛加成对映选择性產物。[129][130][131][132][133]胺与羰化物反应得到的烯胺(类似于烯醇)可作为亲核试剂参与反应,而胺又会从产物脱落重新进入催化循环。烯胺催化法大多基于有机小分子受質,属有机催化。在下图中,脯氨酸对于催化三酮的环化反应非常有效[134][135]:
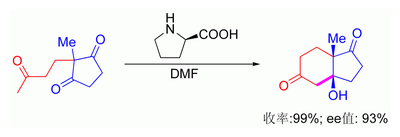
脯氨酸催化的三酮关环反应

该反应也称Hajos-Parrish反应[136][137](还称Hajos-Parrish-Eder-Sauer-Wiechert反应)[138]H-P条件仅需催化量的脯氨酸(3%摩量)。由于过渡态的烯胺中间体比酮(前体)的烯醇式更亲核,因此该反应没有诸如非手性反应的问题。这种策略实用价值很高,因为它提供了简单促成对映选择性反应的方法,而且不需要使用昂贵或有毒的过渡金属催化剂。在2000年BarbasIII和同事在前人的基础改善了该方法,发现30%摩量脯氨酸催化可使羟醛反应對映體過剩99%以上;而羟基丙酮受質则可以得到高选择性的顺式双羟基产物[139][140]。
值得一提的是脯氨酸催化的羟醛反应沒有任何「非线性效应」(线性效应指产物对映选择性的比例直接与催化剂的光学纯度相关)。结合同位素标记得到的证据及计算化学研究,假定的脯氨酸催化的羟醛反应机理如下:[141]
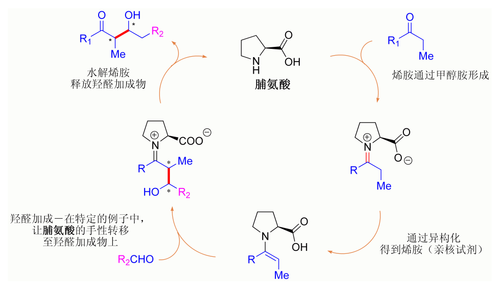
假定的脯氨酸催化的羟醛反应机理

这种策略允许有竞争双醛交叉羟醛反应。通常两醛之间的交叉羟醛反应都有竞争,因此它们很容易聚合或有隨機反应而得到混合产物。第一例如下:[142]

脯氨酸催化的交叉-羟醛反应

基于烯醇的羟醛加成反应会优先形成顺式产物,而有机催化形成的加成产物是选反的。许多实例中,有机催化条件可以非常温以防止多聚化。然而由于两种亲电试剂都能产生烯醇化质子,所以为了提高选择性就必须使用注射泵缓慢滴加亲电试剂加以控制。如果其中一种原料沒有烯醇化的α氢原子或β侧链就可实现可控制的加成反应。
麦克米伦和同事于2004年发现了一个很好的展示不对称有机催化羟醛反应的例子,他们通过不同的方法保护碳水化合物(糖类)。传统合成己糖的方法是使用多步保护、脱保护,共需8至14步完成,而有机催化法能够作用于许多相同受質,且仅用两步就可顺利完成。该反应使用了脯氨酸催化的α羟基醛的二聚反应,然后几步向山羟醛环化反应完成合成。

合成己糖两步法

α羟醛的二聚化反应要防止羟醛加成物繼續加成反应。[143]早期的研究表明能耐受α-烷氧或α-硅氧的保护基适用于此反应,因为能耐受吸电子基团(如乙酰基)的醛都沒反应。保护后的赤藓糖产物通过向山羟醛加成,之后半内缩醛化转化为四种糖分子。这須在向山加成中适当控制非对映体,并且产物硅氧碳正离子会优先环化,而不是繼續羟醛反应,葡萄糖、甘露糖、阿洛糖等都可以此法制备:

构建不同的己糖

“直接”羟醛加成
[编辑]普通的羟醛反应中,羰化物都脫质子形成烯醇。烯醇加入到醛或酮化合物中形成醇盐,然后酸化和后处理。有更好的方法,理论上能够不需多步操作,而可“直接”一步反应,想法是用金属催化剂在羟醛加成过程中释放。而问题在于反应产生的醇盐比原料更碱。产物和金属离子紧密键合,以致阻碍了它与羰基原料继续反应[144][145][120]。

直接羟醛反应法

埃文斯发现了用硅基化羟醛加成物反应的方法,[146][147]如硅试剂三甲基氯硅烷参与反应用于代替金属离子结合醇盐,并允许金属催化剂的循环利用。该法减少了反应步数,降低了试剂消耗,使反应更经济且适用于工业领域。

硅试剂在羟醛反应中的应用

一种最近由Shair发现的仿生合成法,使用β-硫酮酸作为亲核试剂。[148]其中一半酮酸在反应中脱羧。其过程非常类似于通过聚酮合成酶来应用丙二-辅酶A。示例中的手性配体为双噁唑啉配体。有趣的是,芳香族和支链脂肪族醛在此例中无法很好地反应。

Shair合成法

生物的羟醛反应
[编辑]
生物化学中的羟醛反应,如糖酵解反应的第二阶段:1,6-二磷酸果糖分解成3-磷酸甘油醛和二羟基丙酮,该反应是由醛缩酶A催化的逆向羟醛反应[149]。又如:萜类在生物中间体甲瓦龙酸中的合成[150]等。
参见
[编辑]註解
[编辑]- ^ 通常情况下,该反应最好在较低温度下进行。温度过高可能更利于发生消除反应。[1]
- ^ 鮑羅丁觀察到乙醛在酸性環境下會二聚化,形成3-羥基丁醛
- ^ 该反应中,亲核试剂为一硼烯醇分子,该分子由二丁基硼三氟甲磺酸盐 (nBu2BOTf)反应衍生,碱则是N,N-二异丙基乙基胺。硫醚在第二步中用雷尼镍/氢气还原法脱除。
参考文献
[编辑]- ^ Klein, David R. Organic chemistry 4th. Hoboken, NJ: Wiley. December 22, 2020: 1014. ISBN 978-1-119-65959-4. OCLC 1201694230.
- ^ Wurtz, C. A. Sur un aldéhyde-alcool. Bulletin de la Société chimique de Paris. 2nd series. 1872, 17: 436–442 [2016-11-18]. (原始内容存档于2020-05-27).
- ^ Wurtz, Ad. Ueber einen Aldehyd-Alkohol. Journal für Praktische Chemie. 1872-02-09, 5 (1) [2022-07-21]. ISSN 0021-8383. doi:10.1002/prac.18720050148. (原始内容存档于2022-12-07) (德语).
- ^ Wurtz, C. A. Sur un aldéhyde-alcool. Comp. Rend. 1872, 74: 1361 [2011-03-17]. (原始内容存档于2012-01-11).
- ^ Wade, L. G. Organic Chemistry. Upper Saddle River, New Jersey: Prentice Hall. 6th ed. 2005: 1056–1066. ISBN 0-13-236731-9.
- ^ Smith, M. B.; March, J. Advanced Organic Chemistry. New York: Wiley Interscience. 5th ed. 2001: 1218–1223. ISBN 0-471-58589-0.
- ^ Mahrwald, R. Modern Aldol Reactions, Volumes 1 and 2. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA. 2004: 1218–1223. ISBN 3-527-30714-1.
- ^ Louis-François HOLLENDER. CHRONIQUE HISTORIQUE Le strasbourgeois Charles Adolphe Wurtz (1817-1884) Doyen de la Faculté de Médecine de Paris, Président de l’Académie des Sciences et Président de l’Académie de Médecine (PDF). Bulletin de l'Academie Nationale de Medecine. 2010-04-05, 194: p. 847 – 850 [2015-04-08]. (原始内容存档 (PDF)于2016-03-03).
- ^ Gordin, Michael D. Facing the Music: How Original Was Borodin's Chemistry?. Journal of Chemical Education. 2006-04, 83 (4) [2022-07-21]. ISSN 0021-9584. doi:10.1021/ed083p561. (原始内容存档于2022-12-07) (英语).
- ^ Behrman, E. J. Borodin. Journal of Chemical Education. 2006-08, 83 (8) [2022-07-21]. ISSN 0021-9584. doi:10.1021/ed083p1138.1. (原始内容存档于2022-07-21) (英语).
- ^ Rainer Mahrwald. Modern Aldol Reactions. Wiley. 6 August 2004. ISBN 978-3-527-30714-2.
- ^ 12.0 12.1 Zimmerman, Howard E.; Traxler, Marjorie D. The Stereochemistry of the Ivanov and Reformatsky Reactions. I. Journal of the American Chemical Society. 1957-04, 79 (8) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja01565a041. (原始内容存档于2022-07-21) (英语).
- ^ Hoang, Linh; Bahmanyar, S.; Houk, K. N.; List, Benjamin. Kinetic and Stereochemical Evidence for the Involvement of Only One Proline Molecule in the Transition States of Proline-Catalyzed Intra- and Intermolecular Aldol Reactions. Journal of the American Chemical Society. 2003-01-01, 125 (1) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja028634o. (原始内容存档于2022-07-21) (英语).
- ^ Trost, Bm. The Atom Economy—A Search for Synthetic Efficiency. Science. 1991-12-06, 254 (5037) [2022-07-21]. ISSN 0036-8075. doi:10.1126/science.1962206. (原始内容存档于2022-10-09) (英语).
- ^ Reetz, M. T.; Huellmann, M.; Massa, W.; Berger, S.; Rademacher, P.; Heymanns, P. Structure and electronic nature of the benzaldehyde/boron trifluoride adduct. Journal of the American Chemical Society. 1986-04, 108 (9) [2022-07-15]. ISSN 0002-7863. doi:10.1021/ja00269a044. (原始内容存档于2022-07-21) (英语).
- ^ Keck, Gary E.; Castellino, Stephen. On the origins of stereoselectivity in chelation controlled nucleophilic additions to .beta.-alkoxy aldehydes: solution structures of Lewis acid complexes via NMR spectroscopy. Journal of the American Chemical Society. 1986-06, 108 (13) [2022-07-15]. ISSN 0002-7863. doi:10.1021/ja00273a060. (原始内容存档于2022-07-21) (英语).
- ^ Keck, Gary E; Castellino, Stephen. Direct Evidence for the Absence of Chelation with β-Silyloxy Aldehydes and Lewis Acids. Tetrahedron Letters. 1987-01-01, 28 (3). ISSN 0040-4039. doi:10.1016/S0040-4039(00)95707-1 (英语).
- ^ Mukaiyama, Teruaki; Banno, Kazuo; Narasaka, Koichi. New cross-aldol reactions. Reactions of silyl enol ethers with carbonyl compounds activated by titanium tetrachloride. Journal of the American Chemical Society. 1974-11, 96 (24) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00831a019. (原始内容存档于2022-07-21) (英语).
- ^ Mukaiyama, Teruaki; Narasaka, Koichi. 3-Hydroxy-3-methyl-1-phenyl-1-butanone by Crossed Aldol Reaction: 1-Butanone, 3-hydroxy-3-methyl-1-phenyl-. John Wiley & Sons, Inc. (编). Organic Syntheses. Hoboken, NJ, USA: John Wiley & Sons, Inc. 2003-04-28: 6–6 [2022-07-21]. ISBN 978-0-471-26422-4. doi:10.1002/0471264180.os065.02. (原始内容存档于2022-07-21) (英语).
- ^ Mukaiyama, T., Narasaka, K., Banno, K. New aldol type reaction.. Chem. Lett. 1973: 1011–1014.
- ^ Mukaiyama, Teruaki; Banno, Kazuo; Narasaka, Koichi. ChemInform Abstract: NEW CROSS-ALDOL REACTIONS, REACTIONS OF SILYL ENOL ETHERS WITH CARBONYL COMPOUNDS ACTIVATED BY TITANIUM TETRACHLORIDE. Chemischer Informationsdienst. 1975-02-11, 6 (6) [2022-07-21]. doi:10.1002/chin.197506159. (原始内容存档于2022-07-21) (德语).
- ^ Palomo, Claudio; Oiarbide, Mikel; García, Jesús M. Current progress in the asymmetric aldol addition reaction. Chemical Society Reviews. 2004-02-06, 33 (2) [2022-07-21]. ISSN 1460-4744. doi:10.1039/B202901D. (原始内容存档于2022-10-17) (英语).
- ^ 23.0 23.1 Dellaria, J. F. Jun.; Santarsiero, B. D. ChemInform Abstract: Stereoselective Alkylation of Chiral Glycine Enolate Synthons. The Enantioselective Synthesis of α-Amino Acid Derivatives.. ChemInform. 1989-05-30, 20 (22) [2022-07-21]. doi:10.1002/chin.198922293. (原始内容存档于2022-07-21) (英语).
- ^ 24.0 24.1 DIASTEREOSELECTIVE ALDOL CONDENSATION USING A CHIRAL OXAZOLIDINONE AUXILIARY: (2S,3S)-3-HYDROXY-3-PHENYL-2-METHYLPROPANOIC ACID. Organic Syntheses. 1990, 68 [2022-07-21]. doi:10.15227/orgsyn.068.0083. (原始内容存档于2022-06-24).
- ^ Mlynarski, Jacek; Bas, Sebastian. ChemInform Abstract: Catalytic Asymmetric Aldol Reactions in Aqueous Media - A 5 Year Update. ChemInform. 2014-04-28, 45 (17) [2022-07-21]. doi:10.1002/chin.201417261. (原始内容存档于2022-07-21) (英语).
- ^ A. Tanaka and C. F. Barbas, III. in Modern Aldol Reactions, Volume. Wiley-VCH. 2004.
- ^ Bednarski, M. D. Applications of enzymic aldol reactions in organic synthesis. Applied Biocatalysis 1991, 1, 87-116.
- ^ Fessner, W.-D. ChemInform Abstract: Enzyme-Catalyzed Aldol Additions in Asymmetric Synthesis. Part 1. ChemInform. 2010-08-20, 24 (22) [2022-07-21]. doi:10.1002/chin.199322322. (原始内容存档于2022-07-21) (英语).
- ^ Fessner, W. D. Enzyme-catalyzed aldol additions in asymmetric synthesis. Part 2. Kontakte (Darmstadt) 1993, 23-34.
- ^ Petersen, M., Zannetti, M. T., Fessner, W.-D. Tandem asymmetric C-C bond formations by enzyme catalysis. Top. Curr. Chem. 1997, 186, 87-117.
- ^ Petersen, M.; Zannetti, M. T.; Fessner, W.-D. ChemInform Abstract: Tandem Asymmetric C-C Bond Formations by Enzyme Catalysis. ChemInform. 2010-08-03, 28 (28) [2022-07-21]. doi:10.1002/chin.199728223. (原始内容存档于2022-07-21) (英语).
- ^ TAKAYAMA, S.; MCGARVEY, G. J.; WONG, C.-H. ChemInform Abstract: Enzymes in Organic Synthesis: Recent Developments in Aldol Reactions and Glycosylations. ChemInform. 2010-06-23, 29 (14). ISSN 0931-7597. doi:10.1002/chin.199814248.
- ^ W.-D. Fessner, in Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Berlin, 2004, vol. 1, ch. 5, pp. 201–272.
- ^ Gröger, Harald; Vogl, Erasmus M.; Shibasaki, Masakatsu. <1137::aid-chem1137>3.0.co;2-z New Catalytic Concepts for the Asymmetric Aldol Reaction. Chemistry - A European Journal. 1998-07-10, 4 (7). ISSN 0947-6539. doi:10.1002/(sici)1521-3765(19980710)4:7<1137::aid-chem1137>3.0.co;2-z.
- ^ M. Shibasaki, S. Matsunaga and N. Kumagai, in Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Berlin, 2004, vol. 2, ch. 6, pp. 197–227.
- ^ A. Yanagisawa, in Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Berlin, 2004, vol. 2, ch. 1, pp. 1–23.
- ^ Heathcock, C. H. Comp. Org. Syn.. Oxford: Pergamon. 1991: 133–179. ISBN 0-08-040593-2.
- ^ Mukaiyama, Teruaki. The Directed Aldol Reaction. John Wiley & Sons, Inc. (编). Organic Reactions. Hoboken, NJ, USA: John Wiley & Sons, Inc. 1982-08-09: 203–331 [2022-07-21]. ISBN 978-0-471-26418-7. doi:10.1002/0471264180.or028.03. (原始内容存档于2022-12-07) (英语).
- ^ Paterson, I. New Asymmetric Aldol Methodology Using Boron Enolates. Chem. Ind. 1988, 12: 390–394.
- ^ Mestres, Ramon. A green look at the aldol reaction. Green Chemistry. 2004, 6 (12). ISSN 1463-9262. doi:10.1039/b409143b (英语).
- ^ Braun, Manfred; Devant, Ralf. (R)- and (S)-2-acetoxy-1,1,2-triphenylethanol - effective synthetic equivalents of a chiral acetate enolate. Tetrahedron Letters. 1984-01, 25 (44) [2022-07-21]. doi:10.1016/S0040-4039(01)91110-4. (原始内容存档于2020-11-06) (英语).
- ^ Jie Jack Li; et al. Contemporary Drug Synthesis. Wiley-Interscience. 2004: 118. ISBN 0-471-21480-9.
- ^ Schetter, Bernd; Mahrwald, Rainer. Modern Aldol Methods for the Total Synthesis of Polyketides. Angewandte Chemie International Edition. 2006-11-20, 45 (45) [2022-07-21]. doi:10.1002/anie.200602780. (原始内容存档于2022-10-17) (英语).
- ^ Ariëns, E. J. Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology. European Journal of Clinical Pharmacology. 1984, 26 (6) [2022-07-21]. ISSN 0031-6970. doi:10.1007/BF00541922. (原始内容存档于2024-04-13) (英语).
- ^ Journal of the American Chemical Society, 1990 , vol. 112, # 14 p. 5583 - 5601
- ^ Journal of Organic Chemistry, 1996 , vol. 61, # 20 p. 6856 - 6872.
- ^ Nerenberg, J. B.; Hung, D. T.; Somers, P. K.; Schreiber, S. L. J. Am. Chem. Soc. 1993, 115, 12621–12622.
- ^ Harried, S. S.; Yang, G.; Strawn, M. A.; Myles, D. C. J. Org. Chem. 1997, 62, 6098–6099.
- ^ Journal of the American Chemical Society, 1987 , vol. 109, # 9 p. 2821 - 2822.
- ^ <Bioorganic and Medicinal Chemistry Letters, 2007 , vol. 17, # 9 p. 2554 - 2557.
- ^ I. K. Mangion and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3696.
- ^ B. M. Trost, A. Fettes and B. T. Shireman, J. Am. Chem. Soc., 2004, 126, 2660.
- ^ B. M. Trost, M. U. Frederiksen, J. P. N. Papillon, P. E. Harrington, S. Shin and B. T. Shireman, J. Am. Chem. Soc., 2005, 127, 3666.
- ^ P. M. Pihko and A. Erkkila¨ , Tetrahedron Lett., 2003, 44, 7607.
- ^ J. Carpenter, A. B. Northrup, D. Chung, J. J. M. Wiener, S.-G. Kim and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2008, 47, 3568.
- ^ Gennari, C. In Comprehensive Organic Synthesis; Trost, B. M.,Ed.;Pergamon: Oxford, 1993; Vol. 2, Chapter 2.4, p 629.
- ^ Lefour, J.-M.; Loupy, A. Tetrahedron 1978, 34, 2597.
- ^ Loupy, A.; Meyer, G.; Tchoubar, B. Tetrahedron 1978, 34, 1333.
- ^ Murthy, A. S. N.; Bhardwaj, A. P. J. J. Chem. Soc., Perkin Trans 1984, 2, 727.
- ^ Patai, S. The Chemistry of the Carbonyl Group, Wiley, London, 1966;
- ^ Kresge, A.J. Chem. Soc. Rev. 1996, 25, 275;
- ^ Reetz, M. T.; Hu¨ llmann, M.; Seitz, T. Angew. Chem., Int. Ed. Engl. 1987, 26, 477.
- ^ Shambayati, S.; Schreiber, S. L. in Comprehensive Organic Synthesis; Trost, B. M., Ed.; Pergamon: Oxford, 1993; Vol. 1, Chapter 1.10, p 283.
- ^ 64.0 64.1 Gennari, C. In Comprehensive Organic Synthesis; Trost, B. M., Ed.; Pergamon: Oxford, 1993; Vol. 2, Chapter 2.4, p 629.
- ^ Guthrie, J. Peter; Cooper, Kevin J.; Cossar, John; Dawson, Brian A.; Taylor, Kathleen F. The retroaldol reaction of cinnamaldehyde. Canadian Journal of Chemistry. 1984-08-01, 62 (8). ISSN 0008-4042. doi:10.1139/v84-243 (英语).
- ^ Heathcock, C. H. The Aldol Reaction: Group I and II Enolates. in Comp. Org. Synth. (eds. Trost, B. M.,Fleming, I.), 1, 181-231 (Pergamon Press, Oxford, 1991).
- ^ 67.0 67.1 Mukaiyama, T. The directed aldol reaction. Org. React. 1982, 28, 203-331.
- ^ Heathcock, C. H. The Aldol Reaction: Acid and General Base Catalysis. in Comp. Org. Synth. (eds. Trost, B. M.,Fleming, I.), 1, 133-179(Pergamon Press, Oxford, 1991).
- ^ William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
- ^ Heathcock, Clayton H.; Buse, Charles T.; Kleschick, William A.; Pirrung, Michael C.; Sohn, John E.; Lampe, John. Acyclic stereoselection. 7. Stereoselective synthesis of 2-alkyl-3-hydroxy carbonyl compounds by aldol condensation. The Journal of Organic Chemistry. 1980-03, 45 (6) [2022-07-21]. ISSN 0022-3263. doi:10.1021/jo01294a030. (原始内容存档于2022-07-21) (英语).
- ^ 存档副本. [2023-05-02]. (原始内容存档于2023-09-05).
- ^ Palomo, C., Oiarbide, M., Garcia, J. M. The aldol addition reaction: an old transformation at constant rebirth. Chem.-- Eur. J. 2002, 8, 36-44.
- ^ [Tetrahedron, 2002, vol. 58, # 41, p. 8269 - 8280]
- ^ [Tetrahedron Letters, 1997, vol. 38, # 50, p. 8727 - 8730]
- ^ Kinastowski, Stefan; Mroczyk, Wieslawa[Polish Journal of Chemistry, 1984, vol. 58, # 1-3, p. 179 - 184]
- ^ Franklin, A. S., Paterson, I. Recent developments in asymmetric aldol methodology. Contemp. Org. Synth. 1994, 1, 317-338.
- ^ Heathcock, C. H. The aldol condensation as a tool for stereoselective organic synthesis. Curr. Trends Org. Synth., Proc. Int. Conf., 4th 1983, 27-43.
- ^ Knoevenagel, E. Ueber eine Darstellungsweise des Benzylidenacetessigesters. Berichte der deutschen chemischen Gesellschaft. 1896-01, 29 (1). ISSN 0365-9496. doi:10.1002/cber.18960290133 (英语).
- ^ Knoevenagel, E. Condensation von Malonsäure mit aromatischen Aldehyden durch Ammoniak und Amine. Berichte der deutschen chemischen Gesellschaft. 1898-10, 31 (3). ISSN 0365-9496. doi:10.1002/cber.18980310308 (英语).
- ^ House, Herbert O. Modern Synthetic Reactions. Menlo Park, CA.: W. A. Benjamin. 1972. ISBN 978-0-8053-4501-8.
- ^ Bal, B.; Buse, C. T.; Smith, K.; Heathcock, C. H. Org. Syn., Coll. Vol. 7, p.185 (1990); Vol. 63, p.89 (1985). (Article (页面存档备份,存于互联网档案馆))
- ^ Evans, D. A., Nelson, J. V., Taber, T. R. Stereoselective aldol condensations. Top. Stereochem. 1982, 13, 1-115.
- ^ Caminati, Walther; Grabow, Jens-Uwe. The C 2 v Structure of Enolic Acetylacetone. Journal of the American Chemical Society. 2006-01-01, 128 (3) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja055333g. (原始内容存档于2022-07-21) (英语).
- ^ Antonov L. Tautomerism: Methods and Theories 1st. Weinheim: Wiley-VCH. 2013. ISBN 978-3-527-33294-6.
- ^ Smith MB, March J. Advanced Organic Chemistry 5th. New York: Wiley Interscience. 2001: 1218–1223. ISBN 0-471-58589-0.
- ^ Alan R. Katritzky, Elguero J; et al. The Tautomerism of heterocycles. New York: Academic Press. 1976. ISBN 0-12-020651-X.
- ^ Balabin, Roman M. Tautomeric equilibrium and hydrogen shifts in tetrazole and triazoles: Focal-point analysis and ab initio limit. The Journal of Chemical Physics. 2009-10-21, 131 (15) [2022-07-21]. ISSN 0021-9606. doi:10.1063/1.3249968. (原始内容存档于2022-07-21) (英语).
- ^ Hart, H.; Rappoport, Z.; Biali, S.E., in Rappoport, Z. The Chemistry of Enols, Wiley, NY, 1990, pp. 481–589;
- ^ Rappoport, Z.; Biali, S.E. Acc. Chem. Res. 1988, 21, 442.
- ^ Bekker, R.A.; Knunyants, I.L. Sov. Sci. Rev. Sect. B 1984, 5, 145.
- ^ Iglesias, E. J. Org. Chem, 2003, 68, 2680.
- ^ Kresge, A.J. Pure Appl. Chem. 1991, 63, 213; Capon, B., in Rappoport, Z. The Chemistry of Enols, Wiley, NY, 1990, pp. 307–322
- ^ Heathcock, C. H. Science 1981, 214, 395.
- ^ Takacs, J. M.; McGee, L. R.; Ennis, M. D.; Mathre, D. J.; Bartroli, J. Pure Appl. Chem. 1981, 53, 1109
- ^ 95.0 95.1 Brown, Herbert C.; Dhar, Raj K.; Bakshi, Raman K.; Pandiarajan, Paul K.; Singaram, Bakthan. Major effect of the leaving group in dialkylboron chlorides and triflates in controlling the stereospecific conversion of ketones into either [E]- or [Z]-enol borinates. Journal of the American Chemical Society. 1989-04, 111 (9) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00191a058. (原始内容存档于2022-07-21) (英语).
- ^ Braun, M. Methoden Org. Chem. (Houben Weyl), 4th Ed. 1952-1986, E21, 1603.
- ^ Wade, L. G. Organic Chemistry. Upper Saddle River, New Jersey: Prentice Hall. 6th ed. 2005: 1056–1066. ISBN 0-13-236731-9.
- ^ IUPAC Commission on the Nomenclature of Organic Chemistry and Commission on Physical Organic Chemistry, Glossary of Class Names of Organic Compounds and Reactive Intermediates Based on Structure. IUPAC Recommendations 1995. Prepared for publication by G.P. Moss, P.A.S. Smith, D. Tavernier. Pure Appl. Chem. 67, 1307-1375 (1995).
- ^ Paterson, I. Org. React. 1997, 51, 1.
- ^ Ireland, Robert E.; Willard, Alvin K. The stereoselective generation of ester enolates. Tetrahedron Letters. 1975-01, 16 (46) [2022-07-21]. doi:10.1016/S0040-4039(00)91213-9. (原始内容存档于2019-10-13) (英语).
- ^ Narula, Acharan S. An analysis of the diastereomeric transition state interactions for the kinetic deprotonation of acyclic carbonyl derivatives with lithium diisopropylamide. Tetrahedron Letters. 1981-01, 22 (41) [2022-07-21]. doi:10.1016/S0040-4039(01)82081-5. (原始内容存档于2019-09-26) (英语).
- ^ Ireland, Robert E.; Wipf, Peter; Armstrong, Joseph D. Stereochemical control in the ester enolate Claisen rearrangement. 1. Stereoselectivity in silyl ketene acetal formation. The Journal of Organic Chemistry. 1991-01, 56 (2) [2022-07-21]. ISSN 0022-3263. doi:10.1021/jo00002a030. (原始内容存档于2022-07-21) (英语).
- ^ Xie, Linfeng; Isenberger, Kurt M.; Held, Gary; Dahl, Linnea M. Highly Stereoselective Kinetic Enolate Formation: Steric vs Electronic Effects. The Journal of Organic Chemistry. 1997-10-01, 62 (21) [2022-07-21]. ISSN 0022-3263. doi:10.1021/jo971260a. (原始内容存档于2022-07-21) (英语).
- ^ Tetrahedron Volume 76, Issue 4, 24 January 2020, 130618
- ^ Tetrahedron Volume 76, Issue 4, 24 January 2020, 130618
- ^ 106.0 106.1 Cowden, Cameron J.; Paterson, Ian. Asymmetric Aldol Reactions Using Boron Enolates. John Wiley & Sons, Inc. (编). Organic Reactions. Hoboken, NJ, USA: John Wiley & Sons, Inc. 1997-08-28: 1–200 [2022-07-21]. ISBN 978-0-471-26418-7. doi:10.1002/0471264180.or051.01. (原始内容存档于2022-10-24) (英语).
- ^ Evans, D. A.; Nelson, J. V.; Vogel, E.; Taber, T. R. Stereoselective aldol condensations via boron enolates. Journal of the American Chemical Society. 1981-06, 103 (11) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00401a031. (原始内容存档于2022-07-21) (英语).
- ^ Evans, David A.; Rieger, Dale L.; Bilodeau, Mark T.; Urpi, Felix. Stereoselective aldol reactions of chlorotitanium enolates. An efficient method for the assemblage of polypropionate-related synthons. Journal of the American Chemical Society. 1991-01, 113 (3) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00003a051. (原始内容存档于2022-07-21) (英语).
- ^ Evans, D. A. et al. Top. Stereochem. 1982, 13, 1–115. (Review)
- ^ Roush, William R. Concerning the diastereofacial selectivity of the aldol reactions of .alpha.-methyl chiral aldehydes and lithium and boron propionate enolates. The Journal of Organic Chemistry. 1991-06, 56 (13) [2022-07-21]. ISSN 0022-3263. doi:10.1021/jo00013a015. (原始内容存档于2022-07-21) (英语).
- ^ Masamune, Satoru; Ellingboe, John W.; Choy, William. Aldol strategy: coordination of the lithium cation with an alkoxy substituent. Journal of the American Chemical Society. 1982-10, 104 (20) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00384a062. (原始内容存档于2022-07-21) (英语).
- ^ 112.0 112.1 Evans, David A.; Dart, Michael J.; Duffy, Joseph L.; Rieger, Dale L. Double Stereodifferentiating Aldol Reactions. The Documentation of "Partially Matched" Aldol Bond Constructions in the Assemblage of Polypropionate Systems. Journal of the American Chemical Society. 1995-09, 117 (35) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00140a027. (原始内容存档于2022-07-21) (英语).
- ^ Masamune, Satoru; Choy, William; Petersen, John S.; Sita, Lawrence R. Double Asymmetric Synthesis and a New Strategy for Stereochemical Control in Organic Synthesis. Angewandte Chemie International Edition in English. 1985-01, 24 (1) [2022-07-21]. ISSN 0570-0833. doi:10.1002/anie.198500013. (原始内容存档于2022-08-31) (英语).
- ^ Evans, D. A.; Bartroli, J.; Shih, T. L. Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates. Journal of the American Chemical Society. 1981-04, 103 (8) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00398a058. (原始内容存档于2022-07-21) (英语).
- ^ 115.0 115.1 Evans, David A.; Bender, Steven L.; Morris, Joel. The total synthesis of the polyether antibiotic X-206. Journal of the American Chemical Society. 1988-04, 110 (8) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00216a026. (原始内容存档于2022-07-21) (英语).
- ^ Evans, David A.; Clark, J. Stephen; Metternich, Rainer; Novack, Vance J.; Sheppard, George S. Diastereoselective aldol reactions using .beta.-keto imide derived enolates. A versatile approach to the assemblage of polypropionate systems. Journal of the American Chemical Society. 1990-01, 112 (2) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00158a056. (原始内容存档于2022-07-21) (英语).
- ^ Evans, David A.; Ng, Howard P.; Clark, J.Stephen; Rieger, Dale L. Diastereoselective Anti Aldol Reactions of Chiral Ethyl Ketones. Enantioselective Processes for the Synthesis of Polypropionate Natural Products.. Tetrahedron. 1992-01, 48 (11) [2022-07-21]. doi:10.1016/S0040-4020(01)88879-7. (原始内容存档于2019-09-15) (英语).
- ^ Kikukawa, Tadashi; Imaida, Motomasa; Tai, Akira. SYNTHESIS OF THE SEX-ATTRACTANT OF PINE SAWFLIES (DIPRION SPECIES); (2 S ,3 R ,7 R )- AND (2 S ,3 R ,7 S )-3,7-DIMETHYLPENTADECAN-2-OL. Chemistry Letters. 1982-11-05, 11 (11) [2022-07-21]. ISSN 0366-7022. doi:10.1246/cl.1982.1799. (原始内容存档于2022-07-21) (英语).
- ^ Kan, S. B. Jennifer; Ng, Kenneth K.-H.; Paterson, Ian. The Impact of the Mukaiyama Aldol Reaction in Total Synthesis. Angewandte Chemie International Edition. 2013-08-26, 52 (35) [2022-07-21]. doi:10.1002/anie.201303914. (原始内容存档于2022-07-21) (英语).
- ^ 120.0 120.1 Mahrwald, Rainer. Diastereoselection in Lewis-Acid-Mediated Aldol Additions. Chemical Reviews. 1999-05-12, 99 (5) [2022-07-21]. ISSN 0009-2665. doi:10.1021/cr980415r. (原始内容存档于2022-10-26) (英语).
- ^ E. M. Carreira, in Comprehensive Asymmetric Catalysis, Vol. 3, Eds. E.N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Heildelberg, 1999, pp. 997–1065.
- ^ D. Machajewski and C.-H. Wong, Angew. Chem. Int. Ed., 2000, 39, 1352.
- ^ Carreira, Erick M.; Singer, Robert A.; Lee, Wheeseong. Catalytic, Enantioselective Aldol Additions with Methyl and Ethyl Acetate O-Silyl Enolates: A Chiral Tridentate Chelate as a Ligand for Titanium(IV). Journal of the American Chemical Society. 1994-09, 116 (19) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja00098a065. (原始内容存档于2022-07-21) (英语).
- ^ Krüger, Jochen; Carreira, Erick M. Apparent Catalytic Generation of Chiral Metal Enolates: Enantioselective Dienolate Additions to Aldehydes Mediated by Tol-BINAP·Cu(II) Fluoride Complexes. Journal of the American Chemical Society. 1998-02-01, 120 (4) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja973331t. (原始内容存档于2022-08-12) (英语).
- ^ Pagenkopf, Brian L.; Krüger, Jochen; Stojanovic, Aleksandar; Carreira, Erick M. <3124::aid-anie3124>3.0.co;2-1 Mechanistic Insights into Cu-Catalyzed Asymmetric Aldol Reactions: Chemical and Spectroscopic Evidence for a Metalloenolate Intermediate. Angewandte Chemie International Edition. 1998-12-04, 37 (22). ISSN 1433-7851. doi:10.1002/(sici)1521-3773(19981204)37:22<3124::aid-anie3124>3.0.co;2-1.
- ^ Crimmins, Michael T.; King, Bryan W.; Tabet, Elie A. Asymmetric Aldol Additions with Titanium Enolates of Acyloxazolidinethiones: Dependence of Selectivity on Amine Base and Lewis Acid Stoichiometry. Journal of the American Chemical Society. 1997-08-01, 119 (33) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja9716721. (原始内容存档于2022-07-21) (英语).
- ^ Crimmins, Michael T.; Chaudhary, Kleem. Titanium Enolates of Thiazolidinethione Chiral Auxiliaries: Versatile Tools for Asymmetric Aldol Additions. Organic Letters. 2000-03-01, 2 (6) [2022-07-21]. ISSN 1523-7060. doi:10.1021/ol9913901. (原始内容存档于2022-07-21) (英语).
- ^ Crimmins, Org. Lett. 2007, 9(1), 149–152.
- ^ Carreira, Erick M.; Fettes, Alec; MartI, Christiane. Catalytic Enantioselective Aldol Addition Reactions. John Wiley & Sons, Inc. (编). Organic Reactions. Hoboken, NJ, USA: John Wiley & Sons, Inc. 2006-05-23: 1–216 [2022-07-21]. ISBN 978-0-471-26418-7. doi:10.1002/0471264180.or067.01. (原始内容存档于2022-09-18) (英语).
- ^ B. Alcaide and P. Almendros, Angew. Chem., Int. Ed., 2003, 42, 858.
- ^ G. Guillena, C. Najera and D. J. Ramon, Tetrahedron: Asymmetry, 2007, 18, 2249.
- ^ C. Palomo, M. Oiarbide and J. M. Garcia, Chem. Soc. Rev., 2004, 33, 65.
- ^ D. W. C. MacMillan, Nature, 2008, 455, 304.
- ^ C. Pidathala, L. Hoang, N. Vignola and B. List, Angew. Chem., Int. Ed., 2003, 42, 2785.
- ^ C. L. Chandler and B. List, J. Am. Chem. Soc., 2008, 130, 6737.
- ^ Z. G. Hajos, D. R. Parrish, German Patent DE 2102623 1971
- ^ Hajos, Zoltan G.; Parrish, David R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. The Journal of Organic Chemistry. 1974-06, 39 (12) [2022-07-21]. ISSN 0022-3263. doi:10.1021/jo00925a003. (原始内容存档于2021-10-07) (英语).
- ^ Eder, Ulrich; Sauer, Gerhard; Wiechert, Rudolf. New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures. Angewandte Chemie International Edition in English. 1971-07, 10 (7) [2022-07-21]. ISSN 0570-0833. doi:10.1002/anie.197104961. (原始内容存档于2022-07-21) (英语).
- ^ W. Notz and B. List, J. Am. Chem. Soc., 2000, 122, 7386.
- ^ K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260.
- ^ List, Benjamin. The ying and yang of asymmetric aminocatalysis. Chemical Communications. 2006, (8). ISSN 1359-7345. doi:10.1039/b514296m (英语).
- ^ Northrup, Alan B.; MacMillan, David W. C. The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes. Journal of the American Chemical Society. 2002-06-01, 124 (24) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja0262378. (原始内容存档于2022-07-21) (英语).
- ^ Northrup, Alan B.; Mangion, Ian K.; Hettche, Frank; MacMillan, David W. C. Enantioselective Organocatalytic Direct Aldol Reactions ofα-Oxyaldehydes: Step One in a Two-Step Synthesis of Carbohydrates. Angewandte Chemie International Edition. 2004-04-13, 43 (16) [2022-07-21]. ISSN 1433-7851. doi:10.1002/anie.200453716. (原始内容存档于2022-07-21) (英语).
- ^ M. Braun, in Stereoselective Synthesis, Houben-Weyl, Vol. E21/3, Eds. G. Helmchen, R. W. Hoffmann, J. Mulzer and E. Schaumann, Thieme, Stuttgart, 1996, p. 1603.
- ^ C. J. Cowden and I. Paterson, Org. React., 1997, 51, 1.
- ^ Evans, David A.; Tedrow, Jason S.; Shaw, Jared T.; Downey, C. Wade. Diastereoselective Magnesium Halide-Catalyzed anti -Aldol Reactions of Chiral N -Acyloxazolidinones. Journal of the American Chemical Society. 2002-01-01, 124 (3) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja0119548. (原始内容存档于2022-07-21) (英语).
- ^ Evans, David A.; Downey, C. Wade; Shaw, Jared T.; Tedrow, Jason S. Magnesium Halide-Catalyzed Anti-Aldol Reactions of Chiral N -Acylthiazolidinethiones. Organic Letters. 2002-04-01, 4 (7) [2022-07-21]. ISSN 1523-7060. doi:10.1021/ol025553o. (原始内容存档于2022-07-21) (英语).
- ^ Magdziak, Derek; Lalic, Gojko; Lee, Hong Myung; Fortner, Kevin C.; Aloise, Allen D.; Shair, Matthew D. Catalytic Enantioselective Thioester Aldol Reactions That Are Compatible with Protic Functional Groups. Journal of the American Chemical Society. 2005-05-25, 127 (20) [2022-07-21]. ISSN 0002-7863. doi:10.1021/ja051759j. (原始内容存档于2022-07-29) (英语).
- ^ Machajewski, null; Wong, null. The Catalytic Asymmetric Aldol Reaction. Angewandte Chemie (International Ed. in English). 2000-04, 39 (8) [2022-07-21]. ISSN 1521-3773. PMID 10777624. doi:10.1002/(sici)1521-3773(20000417)39:8<1352::aid-anie1352>3.0.co;2-j. (原始内容存档于2022-10-11).
- ^ Brady RO. THE ENZYMATIC SYNTHESIS OF FATTY ACIDS BY ALDOL CONDENSATION. Proceedings of the National Academy of Sciences of the United States of America. 1958;44(10):993-998.
外部連結
[编辑]- Chem 206 Lecture Notes (Fall 2003) by D. A. Evans et al., Harvard University (pp. 345–372)
- Chem 106 Aldol Reaction Notes[永久失效連結]